As of May 10th, 2021 every American 12 years and older is eligible to receive a COVID-19 vaccine. Many still have uncertainties about how these vaccines were developed so fast, deeming them “experimental”. This is probably the most common, and in my opinion understandable question that drives hesitancy towards the COVID-19 vaccines. If typical vaccine development can take 12-15 years1, how could vaccines developed in under 12 months be made without compromising safety? Understanding this requires the context of the history of coronavirus research, vaccine technology development, and the emergency use authorization.
Just How Novel is Coronavirus?
To start, coronaviruses are a common type of virus which causes disease in humans and other animals ranging from mild sickness to deadly infections. Coronaviruses were first formally studied in the 1960s when researchers began to investigate a virus causing what we know as the common cold2. These studies helped us understand how these viruses infect us and other animals in different ways. Because the common cold is, well, common, and because there is such a vast amount of coronaviruses that infect animals, they have been continuously and thoroughly studied4.

The virus that caused the first SARS (severe acute respiratory syndrome) outbreak in 2002 shares 79% of its genome with the virus that causes COVID-19 (SARS-CoV2)5. Not surprisingly, these two viruses use some of the same methods to infect our body6. Because of research on viruses like this, scientists had a huge head-start in understanding the mechanism through which SARS-CoV2 attacks the body as well as how our immune system recognizes the virus. This enabled them to quickly identify the closed conformation of the S2 subunit of the spike protein as the optimal immunological target7,8.
How Were mRNA Vaccines Developed So Fast?

The mRNA vaccine technology underlying COVID vaccines is not all that new, either. The first published research demonstrating lipid nanoparticles as a method to deliver mRNA to cells dates back to 198910, and mRNA vaccines demonstrating immune responses to pathogens were first published in 199411. Since then the excitement of the possibilities that mRNA-based vaccines and treatments bring birthed a spur of biotech start-ups, including Moderna (ModeRNA), whose immense funding quickly helped it reach a valuation of $5 billion long before COVID-19 was in our vocabularies12.
Their big assertion is that mRNA is like software, in that its code can be altered and potentially endless therapeutic developments are possible. In this way, the “software” that is mRNA could be coded to produce the antigen on SARS-CoV2 as soon as its “code” was available. This code became available after the SARS-CoV2 genome was released by Chinese officials on January 10, 202013. On January 15th, 5 days later, mRNA vaccine development began14. Since then, SARS-CoV2 full genomes have been sequenced by labs around the world over 100,000 times as of October 2020 to better understand the virus and track any variants15. Manufacturing mRNA vaccines is exponentially faster, because rather than having to grow trillions of live viruses mRNA simply needs to be assembled from its 4 nucleotide subunits.
How were the adenovirus vaccines developed so fast?
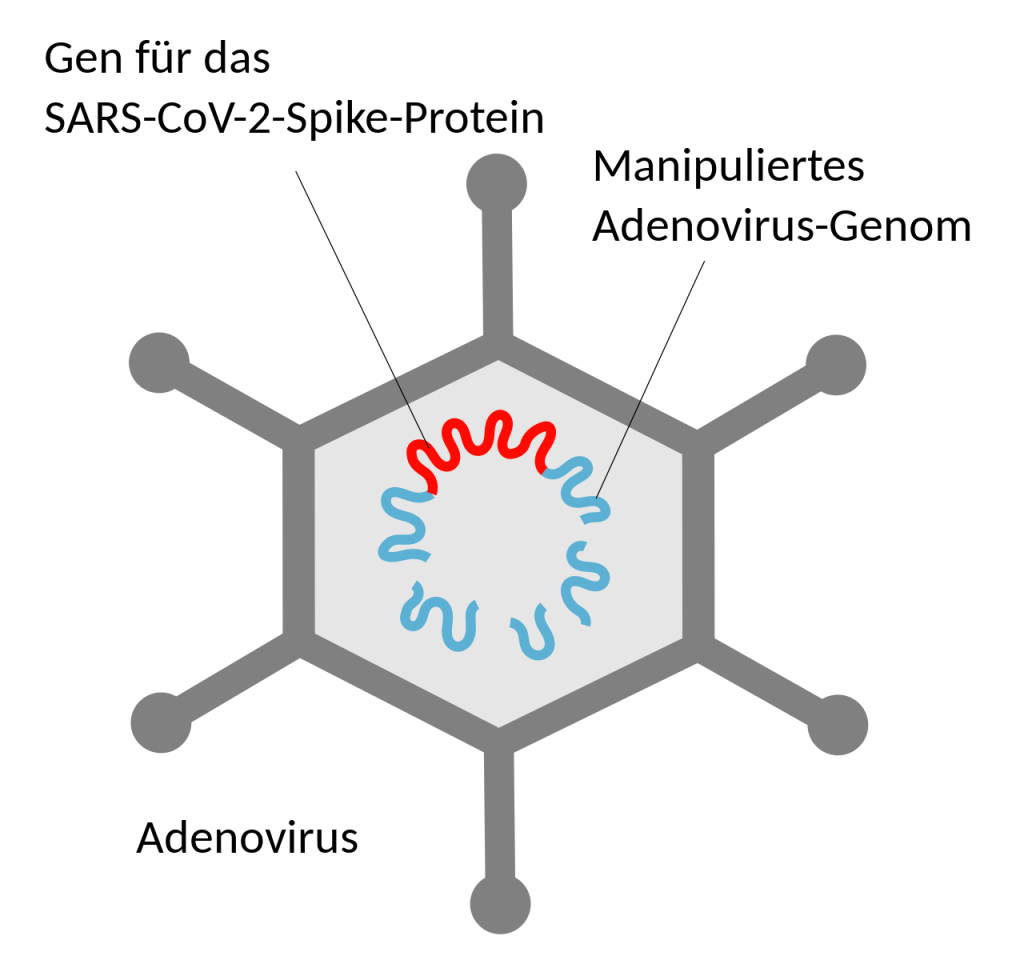
Certain vaccines, including the Johnson and Johnson, AstraZeneca, and Sputnik vaccines, use what is called an adenoviral vector. This vaccine uses a common and harmless adenovirus containing the DNA instructions to make spike protein. This virus is simply used to transport DNA instructions to your cell. It can not infect you because the ability to replicate has been deleted from its genome17. It also lacks the ability to alter our DNA.18
Adenoviral vector technology dates back to the 1990s when they were first being investigated as a method to transfer genetic material to dysfunctional cells causing cystic fibrosis, changing their genetic expression to function normally.19 Adenoviruses cause a strong immune response, which is exactly their challenge – being a common human virus means many people have strong adaptive immune responses which neutralize the virus before they can infect cells. However, this strong immune response proves advantageous for eliciting immune responses to vaccines.20 Later modifications of this technology brought us effective adenoviral vector vaccines, including the ebola virus vaccine which was licensed by the FDA for use in the US in December 2019.21
These initial developments gave vaccine developers an excellent platform using a common and easy to grow virus to induce an immune response against SARS-CoV2 without ever having to touch it. Adenoviral vector vaccines are known to be effective, with immunity that lasts years, and safe, though rare adverse reactions are well documented.23
How Are Vaccines Typically Developed?
Vaccine development usually consists of the following sequential steps, historically taking 12-18 years24:

- Exploratory: Vaccine technology development.
- Pre-Clinical: Animal testing, often on mice or monkeys, to observe immune response and gather early safety data.
- IND (Investigational New Drug Application): Must be approved by the FDA to allow clinical trials.
- Phase I Clinical Trials: A group of 20-80 non-at-risk participants will receive the vaccine. Only if the trial is promising will the research move on to the next step. This tests safety.
- Phase II Clinical Trials: A group of hundreds will receive the vaccine in a randomized, placebo-controlled trial. These may be at-risk individuals with underlying conditions. This tests efficacy.
- Phase III Clinical Trials: A group of tens of thousands will receive the vaccine in another randomized, placebo-controlled trial. This tests safety and efficacy on a large scale.
- Approval and licensure: After a vetting process by the FDA that can take years, the vaccine will be approved for use and given a biologics license.
- Phase IV Clinical Trials: There may even be post-approval clinical trials, as well as adverse event monitoring by the CDC through the VAERS.
This is a back and forth process of research, data collection and analysis, and applications to receive approval to move to the next stage of development. This multi-phase process includes battling bureaucratic government procedures, finding enough participants with interest in participating in a study, and balancing immense cost with possible profits sometimes over a decade away.
How Were COVID-19 Vaccines Developed Faster?

A large body of basic science research has shaped our understanding of coronaviruses, and various vaccine platforms have been developed and improved over decades. The convergence of this foundational knowledge and the global emergency status of the COVID-19 pandemic made development of a vaccine a primary interest for many groups. Researchers, pharmaceutical companies and government bodies across the world almost immediately began development efforts and soon collaborated.
Thanks in part to the $18 billion in funding from Project Warp Speed26, some vaccine developers in the United States could take the financial risk of performing many of the steps of development at the same time. It only makes sense to build extensive manufacturing infrastructure at the same time as clinical trials if there is no risk of losing your own financial investment. Clinical trials can easily not go so well, leading to setbacks or failure. Because there were 6 vaccine projects backed by Project Warp Speed and at least 89 others around the globe27, we saw a few very successful vaccines that led the pack early on, most notably Pfizer-BioNTech and Moderna mRNA vaccines. Only Moderna was funded by Project Warp Speed, though Pfizer also had an initial advance purchase agreement with the US government.28

Due to the fact that COVID-19 has been such a global event that has shaken normal life for nearly everyone, researchers were able to quickly mobilize tens of thousands of volunteers for each trial. The number of volunteers allowed for the development of robust safety profiles. Additionally, an ongoing pandemic with high levels of circulating virus allowed for high-quality estimations of efficacy in a relatively short period of time. Namely, 2 months for the phase III trials.
Despite the large size of the trials, a longer lasting trial still would not have picked up on the rarest of adverse events. Those which occur less than 1 in a million are so rare that they may never be seen until vaccinations are carried out on a large scale. Even then, these adverse reactions are so rare that they are far outweighed by the individual risk of harm or death by COVID-19.29
What is An Emergency Use Authorization?
An emergency use authorization (EUA) was originally devised to allow rapid deployment of treatments in the case of biological warfare as part of the Bioshield Act of 2004 after 9/11.32 An EUA for a vaccine will only be issued in an emergency situation if large-scale phase III trials show compelling safety and efficacy data.33 Safety and efficacy are weighed to determine if the potential risks outweigh the benefit of the vaccine. A rolling review by the ACIP, a board of experts in vaccinology, immunology, virology, public health, pediatrics, internal medicine, infectious diseases, nursing and/or preventive medicine, will occur during the clinical trials. This committee will rigorously review the available data from trials as well as manufacturing protocols to determine of the vaccine is safe and effective. The safety and efficacy is reviewed frequently to maintain EUA status, and may be revoked at any time.34
To receive an EUA, at least 2 months of safety data is required. Using this pathway allows a solution for an emergency to be expedited through the bureaucratic red-tape and brought to the center stage for consideration. Now that the first approved vaccines have 6 months of compelling safety and efficacy data, they are eligible to apply for full FDA approval.
In Summary
Building on decades of basic science research and technological developments, vaccine manufacturers had a huge head-start in development. The global emergency status of COVID-19 allowed for huge financial risks to be taken through overlapping phases of development and manufacturing, as well as government funding. Due to this emergent status, regulatory hurdles for approval were eliminated, while the highest standards of safety and efficacy data were still required. Researchers were able to gather sufficient data through massive volunteer efforts and real-time data collection during an actively spreading pandemic.
Cover photo courtesy of Spencerbdavis35

{kind=link}
{kind=link}
{kind=link}
_E.jpg){kind=link}
.jpg){kind=link}
{kind=link}
Such a well written article 👏. Thank you for the extensive research you’ve done and for sharing it in such a clear explanation. This is so helpful for people to understand.
LikeLiked by 1 person
I’m so glad you found it informative and enjoyed it!
LikeLike